Prevenzione e Trattamento dei Sanguinamenti – Fifth Aland Island Meeting on von Willebrand Disease
Erik Von Willebrand nel 1926 pubblicava sul Finska Lakaresallskskapets Handlingar, in lingua svedese, un articolo con la descrizione di una sindrome emorragica ereditaria che colpiva i componenti sia maschi che femmine di una famiglia delle isole Aland. Egli chiamò questo disordine emorragico Pseudoemofilia. Negli anni a venire eponimicamente verrà chiamato e conosciuto come la malattia di Von Willebrand (VWD). Da quella osservazione sono passati circa 90 anni e molti sono stati i progressi compiuti nella conoscenza dei meccanismi relativi alla patogenesi e nel trattamento della malattia.
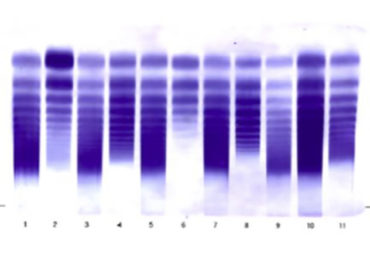
La VWD colpisce più del 1% della popolazione generale rappresentando così il difetto emorragico più diffuso. La VWD è una patologia eterogenea che viene classificata in 3 grandi categorie basandosi sui difetti quantitativi e qualitativi del Fattore di VW (vWF) e dei sui multimeri (fig1)
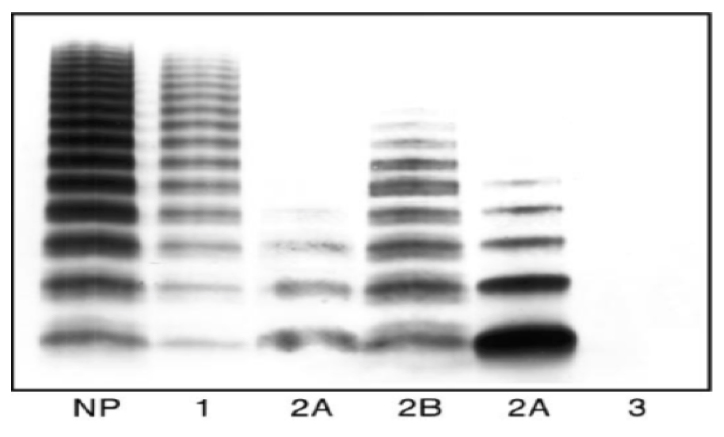
Fig.1 Analisi Multimerica: Plasma normale in confronto con vWD tipo 1, tipo 2 e tipo 3
Il Tipo 1 è la forma più comune e rappresenta il 70/75% dei casi ed caratterizzata dalla presenza di un normale vWF con un deficit parziale solo quantitativo. Il Tipo 2 interessa circa il 20% dei pazienti ed è caratterizzato da un deficit quantitativo in associazione ad alcune modificazioni qualitative del vWF. Il tipo 2 a seconda del deficit qualitativo viene suddiviso in 2A,2B,2M e 2N. Infine il Tipo 3 è rappresentato dal 1-3% dei pazienti ed caratterizzato da una grave carenza o da una assenza totale di vWF che si accompagna a bassi livelli di fattore VIII coagulante (F.VIII:C).
Sulle manifestazioni cliniche, la diagnostica e gli approcci terapeutici molto è stato scritto e pubblicato. Qui vogliamo soffermarci su una manifestazione clinica della WD spesso trascurata dalla letteratura scientifica e che nel corso del Fifth Aland Island meeting on von Willebrand disease 2016 è stata oggetto di particolare attenzione, la Angiodisplasia nella vWD.
La prevenzione ed il trattamento del sanguinamento dovuto alla Angiodisplasia è ancora un importante problema di non facile risoluzione nella gestione dei pazienti con vWD.
L’Angiodisplasia è caratterizzata da lesioni degenerative del sistema vascolare a carico del tratto gastro intestinale della mucosa e associata nel contempo ad un abnorme sviluppo di nuovi vasi (angiogenesi).
I sanguinamenti dovuti a questa condizione sono presenti nella popolazione dei pazienti con una incidenza dal 2.6 a 6.2 %. Il sanguinamento gastrointestinale associato alla angiodisplasia è una complicazione ben documentata in letteratura e si associa quasi esclusivamente a quei pazienti che presentano la mancanza o un grave deficit di Multimeri ad alto peso molecolare (HMWs).
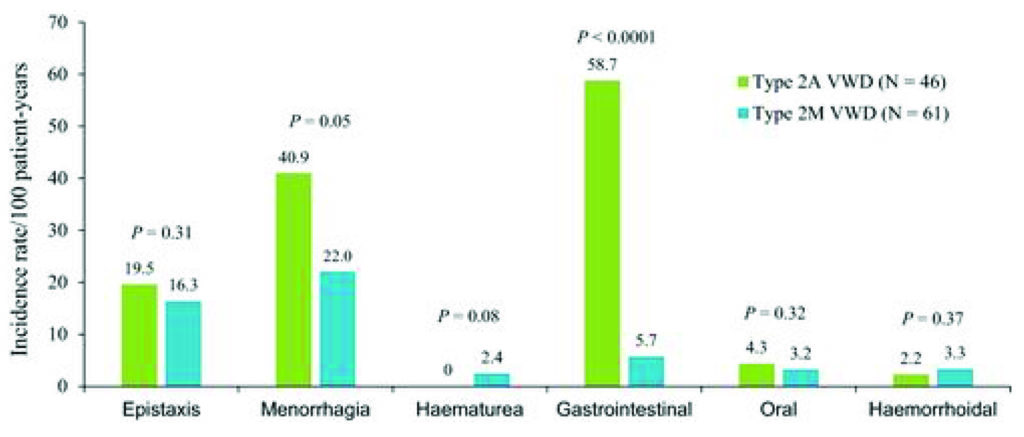
Fig 1 – incidenza 100 pz/anno per sanguinamenti spontanei che hanno richiesto trattamento in pazienti vWD conTipo2A e Tipo2M (Castaman et al. 2012).
Uno studio prospettico della durata di due anni con un arruolamento di 107 pazienti (Castaman G. et al 2012) ha dimostrato come i sanguinamenti GI siano maggiormente presenti, oltre che nei pazienti tipo 3, anche nei pazienti 2A , caratterizzati dalla mancanza di multimeri ad alto peso molecolare, rispetto al Tipo 2M (fig1)
Una convincente dimostrazione indiretta del ruolo dei HMWs nel sanguinamento grastrointestinale dei pazienti con vWD ne viene dalla osservazione dei pazienti con vWD acquisita, in specie nei pazienti con stenosi aortica o con disordini linfoproliferativi. La stenosi aortica è spesso accompagnata da una mancanza di HMWMs a causa dell’elevato shear-stress esercitato dalla valvola stenotica, che modifica la molecola del VWF e ne aumenta la suscettibilità alla proteolisi mediante ADAMTS-13. La correzione chirurgica della stenosi della valvola ripristina un normale pattern multimerico dei HMWMs e abolisce le complicanze emorragiche.
I meccanismi fisiopatologici che sottostanno al sanguinamento gastrointestinale correlato all’angiodisplasia della VWD non è stato ancora del tutto chiarito. Tuttavia, alcuni dati in vitro hanno dimostrato che un basso VWF nei corpuscoli di Weibel- Palade nelle cellule endoteliali può promuovere l’angiogenesi e diminuire la stabilità del vaso e questa condizione potrebbe essere responsabile delle malformazioni vascolari tipiche dell’angiodisplasia.
Il controllo del sanguinamento gastrointestinale in pazienti con VWD e angiodisplasia è di difficile realizzazione a causa delle frequenti recidive e della gravità del sanguinamento che peraltro tende ad aumentare con l’età. La terapia sostitutiva con concentrati di VWF / FVIII derivati dal plasma è il cardine del trattamento per i sanguinamenti gastrointestinali acuti nella VWD. Tuttavia è esperienza diffusa che l’efficacia clinica del VWF / FVIII in questa condizione sia inferiore rispetto all’efficacia registrata in altri sedi sanguinanti. Diverse sono le esperienze che confermano queste osservazioni.
Berntrop E. et al. nel 2009, in uno studio prospettico, ha dimostrato la necessità di una dose giornaliera media più elevata (44 vs 29 UI kg -1) e una durata più lunga del trattamento (4.23 vs 1.93 giorni) per il controllo dei sanguinamenti GI rispetto a quello che era necessario in altre sedi.
Anche Franchini et al. 2014 arriva alle stesse conclusioni nel corso di uno studio in cui si andava a valutare l’efficacia de concentrati di VWF / FVIII nella profilassi secondaria da sanguinamento GI ricorrente.
Anche nel VWD ProphYlaxis Network, un vasto studio sulla profilassi del 2014, è stata valutata retrospettivamente l’efficacia della profilassi secondaria con concentrati di VWF / FVIII in 59 pazienti con VWD clinicamente grave non responsivi ad altri trattamenti. I ricercatori hanno dimostrato che il sanguinamento gastrointestinale ricorrente e l’epistassi sono stati i motivi più frequenti per indurre la profilassi
secondaria, ognuno pari al 23,6% (13/55) dei casi. Sebbene il numero di sanguinamenti gastrointestinali durante la profilassi fosse significativamente ridotto, la percentuale di riduzione della frequenza sanguinante (49%) era inferiore a quella raggiunta per le emorragie articolari (86%).
Una possibile spiegazione della minore efficacia dei concentrati di VWF / FVIII nel trattamento delle emorragie gastrointestinali può essere correlata al fatto che la terapia sostitutiva non ricostituisce il VWF all’interno delle cellule endoteliali e all’interno delle piastrine (solo VWF plasmatico) e questo potrebbe comportare, da parte della composizione dei HMWMs dei concentrati plasmatici, una minore efficacia degli stessi nel trattamento dei sanguinamenti GI rispetto alla loro efficacia in quelli di altri distretti.
In letteratura si può trovare testimonianza di diverse esperienze con diversi alternativi approcci terapeutici per la gestione dei sanguinamenti gastrointestinali ricorrenti. Questi includono gli estrogeni, il progesterone, l’octreotide, l’acido tranexamico e la DDAVP. Tuttavia questi trattamenti, sulla base delle scarse esperienze attualmente disponibili, non sembrano essere efficaci.
Sono stati anche usati farmaci con proprietà anti-angiogeniche, come la talidomide e l’atorvastatina a dosi molto elevate (fino a 80 mg al giorno). Sebbene in alcuni casi si siano riscontrati effetti positivi sulla frequenza del sanguinamento gastrointestinale, sono necessari studi più ampi e controllati per stabilirene l’utilità clinica. Proposti anche approcci chirurgici ma generalmente sono stati ritenuti di difficile praticabilità perché le lesioni angiodisplastiche sono solitamente multiple e diffuse nel tratto GI.
In sintesi, la prevenzione e il trattamento del sanguinamento gastrointestinale dovuto all’angiodisplasia è un problema irrisolto nella gestione dei pazienti VWD. I concentrati di VWF / FVIII rimangono la terapia principale per il trattamento dei sanguinamenti gastrointestinali in tale condizione. Altre strategie, come VWF ricombinante e farmaci anti-angiogenici, potrebbero avere un ruolo futuro nella prevenzione e nel trattamento del sanguinamento gastrointestinale dovuto all’angiodisplasia.
Riferimenti
1. Von Willebrand EA. Hereditär pseudohemofili. Fin Lakaresallsk Handl. 1926;LXVIII:87‐112.
2. Nilsson IM, Blombäck M, Jorpes E, et al. Von Willebrand’s disease and its correction with
human plasma fraction 1‐0. Acta Med Scand. 1957;159:179‐188.
3. Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of
von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb
Haemost. 2006;4:2103‐2114.
4. Federici AB. Clinical and laboratory diagnosis of VWD. Hematology Am Soc Hematol Educ Program. 2014;2014:524‐530.
5. Federici, A. B.; Berntorp, E.; Lee, C. A. “The 80th anniversary of Von Willebrand’s disease: history, management and research”. Haemophilia. 2006 ;12: 563–572.
6. Bentorp E,Agren A,Aledort L,Blomback M,Cnossen MH,Croteau SE,von Depka M,Federici AB,Goodeve A,Goudemand J,Mannucci PM,Mourik M,Onundarson PT,RodeghieroF,Szanto T,Windyga J. Fifth Aland Island conference on von Willebrand disease. Haemophilia. 2018;24s4:5-19.
7. Cramer EM, Caen JP, Drouet L, Breton‐Gorius J. Absence of tubular structures and immunolabeling for von Willebrand factor in the platelet α‐granules from porcine von Willebrand disease. Blood. 1986;68:774‐778.
8. Valentijn KM, Eikenboom J. Weibel‐Palade bodies: a window to von Willebrand disease. J Thromb Haemost. 2013;11:581‐592.
9. Huang RH, Wang Y, Roth R, et al. Assembly of Weibel‐Palade body‐like tubules from N‐ terminal domains of von Willebrand factor. Proc Natl Acad Sci USA. 2008;105:482‐487.
10. Zenner HL, Collinson LM, Michaux G, Cutler DF. High‐pressure freezing provides insights
into Weibel‐Palade body biogenesis. J Cell Sci. 2007;120:2117‐2125.
11. Valentijn KM, Valentijn JA, Jansen KA, Koster AJ. A new look at Weibel‐Palade body
structure in endothelial cells using electron tomography. J Struct Biol. 2008;161:447‐458.
12. Mourik MJ, Faas FG, Valentijn KM, et al. Correlative light microscopy and electron tomography to study Von Willebrand factor exocytosis from vascular endothelial cells.
Methods Cell Biol. 2014;124:71‐92.
13. Mourik MJ, Faas FG, Zimmermann H, et al. Content delivery to newly forming Weibel‐
Palade bodies is facilitated by multiple connections with the Golgi apparatus. Blood.
2015;125:3509‐3516.
14. Mourik MJ, Faas FG, Zimmermann, et al. Towards the imaging of Weibel‐Palade body
biogenesis by serial block face‐scanning electron microscopy. J Microsc. 2015;259:97‐104.
15. Babich V, Meli A, Knipe L, et al. Selective release of molecules from Weibel‐Palade bodies
during a lingering kiss. Blood. 2008;111:5282‐5290.
16. ValentijnKM,SadlerJE,ValentijnJA,etal.FunctionalarchitectureofWeibel‐Paladebodies.
Blood. 2011;117:5033‐5043.
17. Valentijn KM, van Driel LF, Mourik MJ, et al. Multigranular exocytosis of Weibel‐Palade
bodies in vascular endothelial cells. Blood. 2010;116:1807‐1816.
18. Mourik MJ, Valentijn JA, Voorberg J, et al. von Willebrand factor remodeling during
exocytosis from vascular endothelial cells. J Thromb Haemost. 2013;11:2009‐2019.
19. Wang JW, Bouwens EA, Pintao MC, et al. Analysis of the storage and secretion of von Willebrand factor in blood outgrowth endothelial cells derived from patients with von
Willebrand disease. Blood. 2013;121:2762‐2772.
20. Castaman G, Rodeghiero F. The epidemiology of von willebrand disease. In Federici AB,
et al., eds. Von Willebrand Disease: Basic and Clinical Aspects. Hoboken, NJ: John Wiley &
Sons; 2011:86‐90.
21. Mannucci PM, Kyrle PA, Schulman S, et al. Prophylactic efficacy and pharmacokinetically
guided dosing of a von Willebrand factor/factor VIII concentrate in adults and children with von Willebrand’s disease undergoing elective surgery: a pooled and comparative analysis of data from USA and European Union clinical trials. Blood Transfus. 2013;11:533‐540.
22. DiPaolaJ,LethagenS,GillJ,etal.PresurgicalpharmacokineticanalysisofavonWillebrand factor/factor VIII (VWF/FVIII) concentrate in patients with von Willebrand’s disease (VWD) has limited value in dosing for surgery. Haemophilia. 2011;17:752‐758.
23. MakrisM.GastrointestinalbleedinginvonWillebranddisease.ThrombRes.2006;118(Suppl 1):S13‐S17.
24. RandiAM,LaffanMA,StarkeRD.VonWillebrandfactor,angiodysplasiaandangiogenesis. Mediterr J Hematol Infect Dis. 2013;5:e2013060.
25. Franchini M, Mannucci PM. Gastrointestinal angiodysplasia and bleeding in von Willebrand disease. Thromb Haemost. 2014;112:427‐431.
26. Franchini M, Mannucci PM. Von Willebrand disease‐associated angiodysplasia: a few answers, still many questions. Br J Haematol. 2013;161:177‐182.
27. DaneshBJ,SpiliadisC,WilliamsCB,ZambartasCM.Angiodysplasia–anuncommoncause of colonic bleeding: colonoscopic evaluation of 1,050 patients with rectal bleeding and anaemia. Int J Colorectal Dis. 1987;2:218‐222.
28. Sharma R, Gorbien MJ. Angiodysplasia and lower gastrointestinal tract bleeding in elderly patients. Arch Intern Med. 1995;155:807‐812.
29. Fressinaud E, Meyer D. International survey of patients with von Willebrand disease and angiodysplasia. Thromb Haemost. 1993;70:546.
30. Castaman G, Federici AB, Tosetto A, et al. Different bleeding risk in type 2A and 2M von Willebrand disease: a 2‐year prospective study in 107 patients. J Thromb Haemost. 2012;10:632‐638.
31. Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. 2000;84:345‐349.
32. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349:343‐349.
33. SadlerJE.Aorticstenosis,vonWillebrandfactor,andbleeding.NEnglJMed.2003;349:323‐ 325.
34. Panzer S, Badr Eslam R, Schneller A, et al. Loss of high‐molecular‐weight von Willebrand factor multimers mainly affects platelet aggregation in patients with aortic stenosis. Thromb Haemost. 2010;103:408‐414.
35. Starke RD, Ferraro F, Paschalaki KE, et al. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071‐1080.
36. Berntorp E, Windyga J, European Wilate® Study Group. Treatment and prevention of acute bleedings in von Willebrand disease – efficacy and safety of Wilate®, a new generation von Willebrand factor/factor VIII concentrate. Haemophilia. 2009;15:122‐130.
37. AbshireTC,FedericiAB,AlvarezMT,etal.ProphylaxisinsevereformsofvonWillebrand’s disease: results from the von Willebrand Disease Prophylaxis Network (VWD PN). Haemophilia. 2013;19:76‐81.
38. Mannucci PM, Tenconi PM, Castaman G, Rodeghiero F. Comparison of four virus‐ inactivated plasma concentrates for treatment of severe von Willebrand disease: a cross‐over randomized trial. Blood. 1992;79:3130‐3137.
39. Smith SK. Angiogenesis and reproduction. BJOG. 2001;108:777‐783.
40. Somanath PR, Malinin NL, Byzova TV. Cooperation between integrin αvβ3 and VEGFR2 in
angiogenesis. Angiogenesis. 2009;12:177‐185.