
L’emofilia A acquisita (EAA) è una malattia autoimmune causata dalla produzione spontanea di autoanticorpi neutralizzanti, immunoglobuline G (IgG) (inibitori), che hanno come bersaglio il fattore VIII (FVIII) endogeno. L’EAA è verosimilmente sotto-diagnosticata e mal-diagnosticata nella pratica clinica, tale che pochi dati sono disponibili per elaborare linee guida sulla gestione dell’emorragie e l’eradicazione degli anticorpi che causano la malattia. Oggi le domande chiave più importanti sono riassunte come segue:
– aggiornamento sui dati internazionali
– approfondimenti su eziologia e fisiopatologia
– innovazioni terapeutiche
Dati Epidemiologici
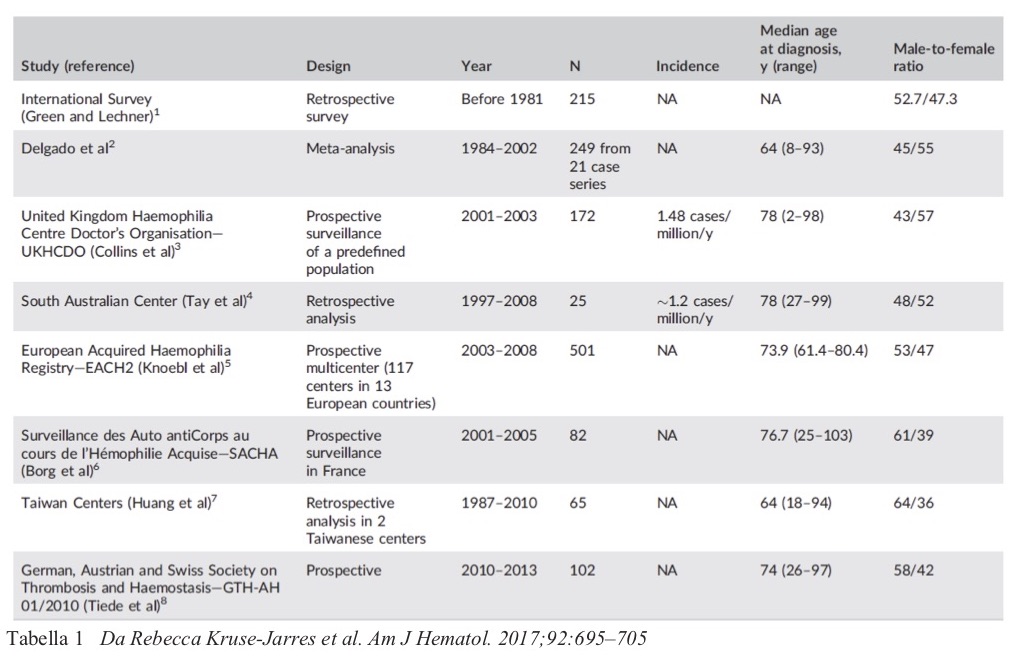
In una recente pubblicazione sono stati riportati i maggiori studi di popolazione che hanno fornito informazioni sulla comprensione dell’AHA fino ad oggi per un totale di 1.412 pazienti (Am J Hematol., 2017; 92: 695-705). (Tabella 1)
L’EAA deve essere presa in considerazione nei pazienti particolarmente anziani e nelle donne nel periodo peripartum e postpartum, con esordio caratterizzato da un sanguinamento anomalo e un prolungamento isolato del tempo di tromboplastina parziale attivata (aPTT) con tempo di protrombina normale (PT). Tuttavia, dovrebbe essere anche sospettato in un paziente asintomatico non in terapia anticoagulante con un aPTT prolungato isolato, un test di miscelazione compatibile con un inibitore e la ricerca del lupus anticoagulante negativa (LA).
Una review ha riportato 42 casi in bambini, inclusi 6 con passaggio transplacentare degli anticorpi materni (Franchini M et al. Pediatr Blood Cancer. 2010; 55: 606-611).
Di seguito viene fornito un algoritmo semplificato per la diagnosi di EAA:
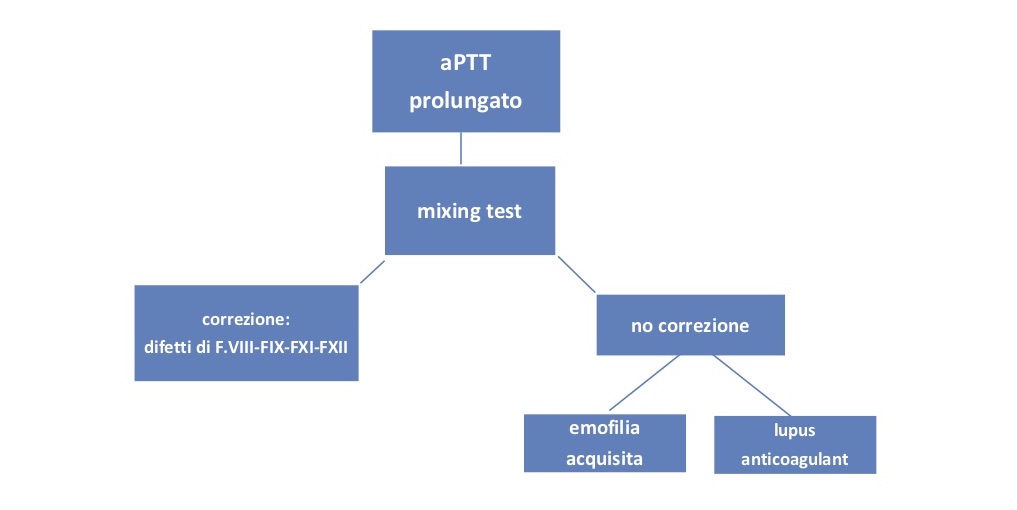
Un inibitore del FVIII è comunemente confermato e quantificato con il test Bethesda o di Nijmegen Bethesda. A causa dell’inattivazione non lineare del FVIII e dell’attività residua dello stesso FVIII entrambi i test possono sottovalutare il titolo dell’inibitore.
La gestione dell’EAA si concentra sui seguenti punti critici:
– Controllo e prevenzione dell’emorragia
– Eradicazione dell’inibitore
– Trattamento della malattia sottostante (ove applicabile)
I casi possono essere:
– idiopatici (43,6% -51,9%)
– associati a malignità (6,4% -18,4%)
– associati a malattie autoimmuni (9,4% -17,0%)
Anche se circa il 30% dei pazienti non richiede un trattamento emostatico, la gravità delle manifestazioni emorragiche varia e potrebbe rappresentare un elevato rischio per la vita.
I tipi e la prevalenza dell’emorragie nell’EAA sono riportati sotto:
– Emorragia sottocutanea (> 80%)
– Sanguinamento gastrointestinale (> 20%)
– Sanguinamento genito-urinario, retroperitoneale e in altri siti (<10%)
– Emorragia intracranica (rara ma potenzialmente fatale)
– Emorragia intraarticolare (rara)
In uno studio il 94,6% dei pazienti presentava emorragia: 77% spontanee e 70% gravi (emoglobina <8 g / dL o diminuzione di Hb> 2 g / dL). (Knoebl P et al. J Thromb Haemost, 2012; 10: 622-631).
Stima della mortalità nell’EAA:
– 20% dei pazienti> 65 anni e con neoplasie sottostanti
– 46% per la malattia di base
– > 38% la causa di morte sconosciuta, che riflette un follow-up limitato nella maggior parte degli studi (Bitting RL et al. Blood Coagul Fibrinol., 2009; 20: 517-523).
Risultati dei dati epidemiologici sulle morti per emorragie:
– 3,2% dei pazienti (17,2% di tutti i decessi) nel registro EACH2
– 9,1% nello studio osservazionale UKHCDO con una mediana di 19 giorni dopo l’esordio dell’EAA
Per quanto la terapia immunosoppressiva sia raccomandata per eliminare gli anticorpi e ridurre il rischio di sanguinamento, essa è associata alla mortalità, che rappresenta il 16% di tutti i decessi (4,2% dei pazienti) nell’EACH2.
A causa della cinetica del secondo ordine degli anticorpi anti-FVIII, i livelli di FVIII non sono predittivi del rischio di sanguinamento e i pazienti possono avere emorragie clinicamente significative nonostante i livelli di attività di FVIII modestamente ridotti (Delgado J et al. Br J Haematol. 2003;121:21–35).
Eziologia e patofisiologia
Una combinazione di fattori genetici e ambientali, insieme all’invecchiamento del sistema immunitario negli anziani, potrebbe portare a una rottura della tolleranza immunitaria, causando lo sviluppo di autoanticorpi contro il FVIII.
Sia l’antigene leucocitario umano (HLA) che l’antigene 4 dei linfociti T citotossici (CTLA-4) svolgono un ruolo importante nel mantenimento della tolleranza delle cellule T periferiche. l’antigene leucocitario umano (HLA) e il CTLA-4 svolgono un ruolo importante nel mantenimento della tolleranza delle cellule T periferiche. È stata osservata una più alta frequenza degli alleli HLA di classe II e dei polimorfismi a singolo nucleotide del gene CTLA-4 in alcune malattie autoimmuni e nell’emofilia A grave. Alleli HLA di classe II con frequenza significativamente più elevata di DRB * 16 [odds ratio (OR) 10.2] e DQB1 * 0502 (OR 2,5) sono stati osservati in pazienti con EAA rispetto ai controlli. Al contrario, gli alleli DRB1 * 15 e DQB * 0602 sono stati rilevati con una frequenza più bassa (OR 0,4 per entrambi i loci HLA) nella stessa coorte. Gli alleli (DRB1 * 16 e DQB1 * 0502), che sono ad alto rischio nell’EAA si rivelano invece a basso rischio nei pazienti affetti da emofilia congenita A con inibitori. Viceversa, gli alleli a basso rischio nell’EAA (DRB1 * 15 e DQB1 * 0602) sono alleli ad alto rischio in pazienti con emofilia A e inibitori. (Tabella 2)
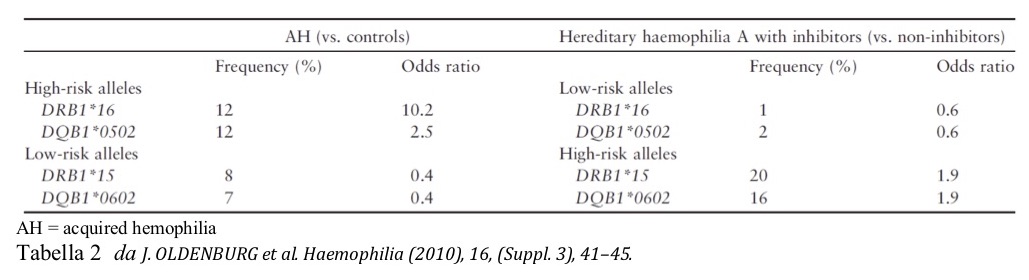
L’allele CTLA-4 + 49 G è stato riscontrato con frequenza relativamente alta nella stessa coorte di pazienti con EAA (OR 2.17), particolarmente nelle donne. I dati indicano che i geni deputati alla risposta immunitaria possono svolgere un ruolo nella formazione di autoanticorpi anti-FVIII nell’EAA. In particolare, livelli più elevati degli alleli HLA di classe II DRB * 16 e DQB1 * 0502 e dell’allele CTLA-4 + 49 G sono stati osservati più frequentemente in pazienti con EAA rispetto ai controlli (Oldenburg et al. Haemophilia (2010), 16, (Suppl. 3), 41-45).
Autoanticorpi anti-FVIII del tipo IgA e non IgG sono predittivi di ricorrenza e di outcome clinico peggiore in pazienti con EAA (Andreas Tiede et al. Blood. 2016;127(19):2289-2297).
Terapia
Le opzioni terapeutiche emostatiche di prima linea sono sintetizzate nella tabella 3.

La terapia sostitutiva con FVIII umano (hFVIII) non è efficace in presenza di inibitori ad alto titolo. Il FVIII ricombinante di origine porcina (rpFVIII), laddove disponibile, rappresenta una novità nel trattamento dei pazienti con EAA, pur essendo già stato utilizzato come plasmaderivato negli emofilici congeniti con inibitori. Esso può raggiungere livelli misurabili di FVIII nei pazienti con EAA, anche se l’inibitore umano è elevato. Per prevedere l’efficacia dei concentrati di hFVIII o di rpFVIII nel trattamento dell’EAA, è essenziale determinare le concentrazioni basali di anticorpi anti-hFVIII e anti-rpFVIII.
L’uso della desmopressina (DDVAP) sottocute può essere riservato ad emorragie minori in pazienti con bassi titoli di inibitore (<2 UB / ml) e livelli di FVIII> 5 UI.L’acido tranexamico in combinazione con l’aPCC o il rFVIIa contribuisce a normalizzare la stabilità del coagulo.
Eradicazione dell’inibitore
La terapia immunosoppressiva (TIS) rappresenta l’intervento più importante per eradicare l’inibitore. Nella tabella 4 è sintetizzata la TIS di prima linea.

La TIS è raccomandata per tutti gli adulti con EAA. Il ruolo della TIS nei pazienti pediatrici non è ben definito a causa della mancanza di dati.
Le immunoglobuline per via endovenosa (IVIG) hanno un ruolo limitato nel trattamento dell’EAA. Nello studio EACH2, l’aggiunta di IVIG ad altri agenti immunosoppressivi come terapia di prima linea non ha fornito alcun beneficio. Un risultato simile è stato osservato nello studio di sorveglianza UKHCDO e in una successiva revisione della letteratura.
La TIS di seconda linea
I pazienti che non riescono ad ottenere un calo del titolo inibitore o un aumento del livello di FVIII al basale dopo 3-5 settimane di TIS di prima linea devono essere considerati per la terapia di seconda linea.
Sebbene le prove siano limitate, le opzioni ragionevoli di seconda linea includono:
- inibitori della calcineurina,
- micofenolato mofetile,
- agenti immunosoppressivi multipli,
- tolleranza immunitaria
- rituximab associato ad altri agenti immunosoppressivi
Non sono disponibili dati sufficienti per raccomandare per l’EAA trattamenti specifici di seconda linea e di linea successiva.
Lo specialista ematologo, che abbia familiarità con l’assistenza dei pazienti con inibitori della coagulazione, con accesso agli agenti emostatici e con un frequente monitoraggio di FVIII, è il più adatto a gestire i pazienti con EAA. La gestione è ottimizzata se è effettuata in stretta collaborazione multispecialistica con altri medici esperti in EAA.